
Review by Abigail Stanton (MOL, G1)
The cell can be a chaotic place to work. Protein employees of all different types rush from room to room, delivering messages, building needed materials, and working together to keep the cell running smoothly. To learn how any one of these proteins does its job, researchers have to consider how they will structure their experiment to get the type of information that they need. One approach is observing the protein at work: what does it do on a normal day? How does it interact with its coworkers? Studying a protein in situ (in its original place) gives researchers the best sense of how the protein actually behaves. However, the complex environment of the cell can make it difficult to pick out the contributions of any one protein. To gain more detailed information, the researcher may need to sit the protein down for a one-on-one interview, purifying it away from the other components of the cell for in vitro (in a test tube) experiments. However, a protein’s behavior alone may be very different from how it acts surrounded by a crowd of molecules. To create the most useful experiment possible, researchers need to find ways to combine the context of in situ studies with the detail and experimental control of in vitro work.
In a recent publication in PNAS, Xia Yao and Xiao Fan, researchers in the lab of Dr. Nieng Yan in the Princeton Department of Molecular Biology, present a new approach for studying protein structures in a more cell-like environment. The Yan lab uses the power of cryo-electron microscopy (cryo-EM) to study proteins that embed themselves in membranes, the molecular walls that form compartments within cells and serve as a barrier between the inside and outside of the cell. While most structural methods take these proteins out of their normal membrane environment, the novel approach developed by Yao et al. allows for study of these proteins in a membrane bubble similar to what they would experience in a cell.
It’s tricky to study a membrane protein
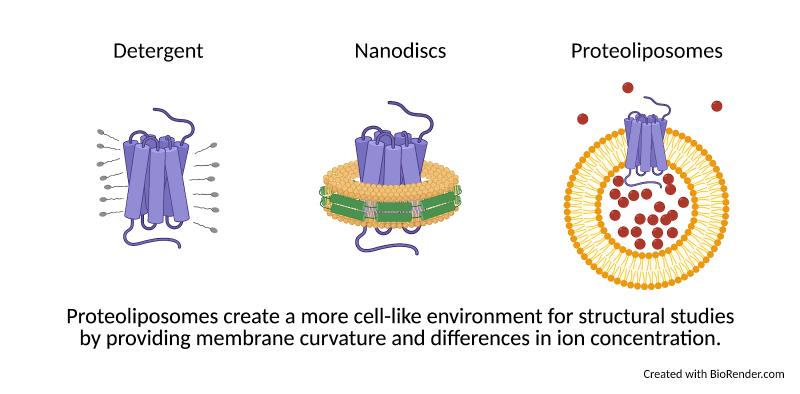
Membrane proteins serve as gates in the membrane walls, communicating signals between the inside and outside of the cell and determining which molecules are allowed to enter or exit. Despite their importance, membrane proteins are notoriously difficult to purify and study. Membranes are maintained by the same force that causes water and oil to separate: hydrophobicity. To keep a purified membrane protein happy, researchers can use detergents (small molecules that have both “oil-like” (hydrophobic) and “water-like” (hydrophilic) regions). These detergent molecules surround the hydrophobic protein and allow it to mix more easily with a watery environment, much like how dish soap helps wash oil off of a pan. However, how a protein looks surrounded by detergent may be different from what it looks like in a membrane. For this reason, researchers also study the structures of proteins in nanodiscs, disk-shaped patches of membrane held together by a protein “belt” (Fig. 1).
Nanodiscs provide a more accurate environment than detergents, but they still lack a few key elements that a protein would normally encounter in the cell. First, while nanodiscs are flat and constricted by their protein belt, biological membranes are flexible and often take on curved shapes that can impact how a protein behaves. Second, membranes divide the cell into compartments, meaning that the concentration of ions on one side of the membrane may be very different than on the other. Ions are small charged molecules that serve as cellular signals and aid in protein function. Like Black Friday shoppers, ions can be packed in on one side of a membrane, ready to push through into the empty store as soon as the door opens. Some membrane proteins may open the doors only when the difference between inside and outside is great enough. Other proteins can harness the force of moving ions, like a water wheel, to make energy for the cell. These activities can’t be studied in nanodiscs, where the ion concentration is the same on all sides.
A new approach from Yao et al.
To study how these forces affect the shape of a protein, Yao et al. developed a novel approach to studying membrane proteins: applying cryo-EM structural techniques to proteins embedded in bubbles of membrane called proteoliposomes. Proteoliposomes form naturally when detergent is slowly removed from a mixture of protein and lipids, the building blocks of membranes. The curvature of proteoliposomes can be adjusted by creating spheres of various sizes, allowing researchers to choose a level of curvature that mimics what the protein would encounter in the cell. In addition, because liposomes create closed “bubbles”, researchers can create an environment inside the bubble that is different from the environment outside. Proteins incorporated into these liposomes can therefore experience membrane curvature and differences of ion concentration like they would inside the cell, much more closely mimicking the “workplace” environment of the protein compared to detergent or nanodiscs.
Proteoliposomes have been used extensively to study protein function, but they are challenging to study on a structural level. Researchers solve high-resolution, cryo-EM protein structures by averaging together thousands of images of a protein to learn what it looks like from every angle and remove background noise. Proteoliposomes form in a broad range of sizes, making it nearly impossible to average them together. To solve this problem, Yao et al. used a technique called size exclusion chromatography to filter their proteoliposomes, ensuring that each would be the same size. Next, proteoliposomes have a tendency to pack in close together on traditional EM grids, making it difficult to pick out individual proteins. Therefore, Yao et al. instead used grids covered with a thin film of well-ordered carbon called graphene. This graphene allows the sample to spread evenly across the grid without adding a lot of background noise. Finally, one of the largest challenges of working with proteoliposomes is the data processing, which involves computationally averaging the individual images together into a final structure. When processing their data, Yao et al. used a deep 2D classification approach, sorting their images multiple times to remove “junk” like images of liposomes with no protein inside.
As a test of their structural workflow, Yao et al. used a bacterial protein called AcrB which acts as a pump to remove unwanted substances from the cell, including antibiotic drugs. The structure of AcrB has been studied many times, making it an excellent reference to make sure the structure produced by their method was accurate. Using their proteoliposome approach, Yao et al. solved the AcrB structure to a resolution of 3.9 Angstrom, comparable to what can be achieved with other structural methods. A key advantage of this approach is that the membrane remains visible in the final structure, allowing researchers to see how the protein is oriented in the liposome. In the case of AcrB, they found that in the vast majority of images, the protein pointed towards the inside of the liposome bubble. This information could be important for correcting functional studies, where it’s assumed that the protein has an equal chance of pointing inwards or outwards.
This work by Yao et al. will open the door to understanding membrane proteins in context and at a level of detail never before achieved. This approach will be particularly informative for proteins that change shape in response to differences in membrane curvature and ion concentration. However, there are still some limitations of this approach that need to be addressed before it can be applied more broadly. First, to see where in the liposome the protein is, a section of the protein needs to stick out of the membrane. Proteins that lack this kind of visibility may need to be tagged or bound to another protein to increase its size. Another challenge is that liposomes can often be “leaky.” Ions, being very small, can slip through gaps in the membrane, removing any difference between inside and outside of the liposome. It will take more work carefully adjusting the conditions of proteoliposome formation to perform true measurements of the effects of ion concentration differences.
Ideally, technical advances will one day allow for researchers to study proteins within the cell with the same level of precision as with purified proteins. Until then, this work by Yao et al. is an important step towards understanding how crucial cellular channels and transporters function. The information gained from these structures will help researchers understand what goes wrong with disease-causing protein mutations and design drugs to help with a broad range of health concerns including pain, cardiovascular disease, and neurological disorders.
Dr. Yan’s research team is eager to apply this new technique. “With this system established, we aim to introduce transmembrane electrochemical potential to study the impact of ion gradients or electric fields on the structure and function of our target proteins,” said Dr. Yan. “This system can also be applied to study multiple cellular events, such as membrane fusion, fission, secretion, etc, at atomic resolutions.”
This original article was published in Proceedings of the National Academy of Sciences on August 4, 2020. Please follow this link to view the full version.
This work is supported by the Dean for Research Innovation Fund for New Ideas in the Natural Sciences from Princeton University. N.Y. is supported by the Shirley M. Tilghman endowed professorship from Princeton University. X. F. is supported by the HFSP long-term fellowship (LT000754) from the International Human Frontier Science Program Organization (HFSPO).