
Review written by Sara Geraghty (QCB, G3)
Proteins are ubiquitous in our cells. Currently, it’s estimated that the human body contains between 80,000 and 400,000 protein molecules, all busily performing the many tasks that keep your cells running smoothly and ultimately go into making you. Not only do proteins form the structural framework of your cells, but they also protect your body against foreign pathogens, help digest your food, and send and transmit signals around your body. However, they don’t do this in isolation: proteins are constantly working hand-in-hand with other proteins and molecules in your body, like your DNA, RNA, small molecules, and ions. When your DNA is replicated, or an ion is actively transported, the proteins doing the job need to recognize those molecules (and often bind to them) in order to carry out their task properly. This fundamental process of a protein binding to a partner molecule, or ligand, is critically important in biological processes ranging from development to cancer. And yet, we know surprisingly little about what proteins bind what ligands, and where — knowledge that is key in understanding, and possibly manipulating, the inner workings of cells.
One important way we currently think about proteins and their interactions is through the study of protein domains. Protein domains are recurring protein subunits (stretches of the amino acid chain) that are grouped into ‘families’. These families share the same or similar amino acid sequence, secondary structure (or the local 3D-folding of certain segments of those amino acids), evolutionary descent, and function. Proteins can vary in the number and types of domains they possess, but the functions of a proteins’ domains come together to form the overall role of a given protein.
The functions of many protein domain families, such as catalyzing a particular reaction or binding DNA to incite transcription, have already been discovered through experiments in the lab. Since many different proteins may share the same domain or domain family, sharing domain family functional annotations can be incredibly useful to biologists when it comes to unraveling the functions of whole proteins. For example, if we have an unknown protein with a protein domain that we know binds DNA, it can be safe to assume that this protein binds DNA (even if we’ve never observed it doing so). Since it is difficult to observe protein function in vivo, most human proteins have been functionally annotated using shared protein domains and domain families.
Though useful, annotating domains using wet lab techniques has some limitations. When it comes to inferring whether a protein domain binds potential ligands, like DNA or RNA, we currently rely on crystal structures. Not only is protein crystallization a laborious process in itself, but in order to figure out if a protein domain binds a particular ligand, we need a specific type of crystal structure: the crystal structure of a protein with that protein domain bound to that particular ligand. This is challenging: just because a protein domain may bind a ligand like DNA, doesn’t mean that it is always bound to DNA. When we crystallize a protein, it won’t necessarily be bound to its partner molecule at the time when it is crystallized. As a result, only one third of all human protein domains have a crystal structure that happens to be bound to any sort of ligand. As an even further complication, even if we do have a crystal structure where the protein is bound to, say, DNA, that does not exclude the possibility that, in other situations, it may also bind RNA, ions, small molecules, or peptides. As a result, we’re left with a majority of protein domains labeled as ‘domains of unknown function’, or labeled with potentially incomplete functional descriptions. This massive knowledge gap is a significant impediment to understanding the full range of functions and interactions a given protein might have, which in turn sets the bound on our understanding of cellular mechanisms.
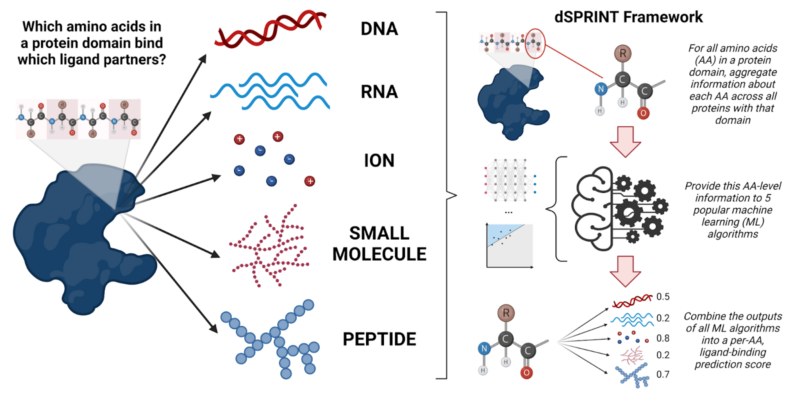
Recently, a study led by graduate student Anat Etzion-Fuchs of Mona Singh’s lab at Princeton University used machine learning techniques to fill this knowledge gap. Etzion-Fuchs et al. created a tool called dSPRINT (domain Sequence-based PRediction of INTeraction-sites). For all protein domains in the human genome, dSPRINT makes predictions about what ligands they bind. It even makes predictions about the specific amino acids in the domain that participate in the interaction. Though the tool is trained using the co-complex (protein bound to ligand) crystal structures that we do have, along with other diverse features, it can be used to make predictions about ligand-binding given an amino acid sequence alone. Since, at present, we only have co-complex crystal structures for about 13% of human proteins, this dramatically expands our ability to describe what exactly these proteins bind to.
Briefly, the tool works by bringing together information across proteins, at the level of the amino acid, for all amino acids within a given protein domain. In other words, each amino acid in the domain gets its own unique profile, complete with many of its distinctive qualities. These qualities include: its physiochemical properties (its acidity, pH, etc.); its predicted secondary structure; to what degree it is preserved across all instances of this domain in proteins with a shared evolutionary history; its allelic frequency across populations (what percentage of the population has this amino acid in this particular position); and more. Thanks to high-throughput sequencing, we have this kind of information for every amino acid, even if we don’t know specifically what a given amino acid binds to. Like a sleuth looking for the telltale signs of a criminal, Etzion-Fuchs et al. wanted to see if they could identify patterns in this data: using the profiles of amino acids that we know bind particular ligand partners, can we pick out other amino acids with similar profiles that also bind the same thing?
To do this, Etzion-Fuchs et al., fed all these amino acid ‘profiles’ into five popular machine learning classifier algorithms: neural networks, random forests, logistic regression, gradient boosting, and support vector machines. They then ‘pasted’ the results of each classifier back together using a novel algorithm, so that they ended up with one final ‘binding score’ per amino acid-ligand combination. Though these five machine learning algorithms are all widely used, they each have their own unique advantages and disadvantages. Using all of these different algorithms together, a technique known as “stacking”, gave better protein-ligand binding predictions than using any of the individual models on their own.
After training their model, Etzion-Fuchs et al. tested its performance using cross-validation — a technique for testing a model on a held out set of data that wasn’t used for training. In other words, they evaluated how well the model worked by seeing how accurately it could predict something that they already knew. This showed that their model can predict known domain-ligand interaction positions from crystal structures with excellent accuracy. From there, they applied their model to the 4,286 human domains for which ligand-binding information is entirely unknown. For each amino acid position within these domains, the model produced a score representing a probability that the amino acid binds DNA, RNA, peptide, small molecule, and ion ligands. Many of dSPRINT’s predictions are consistent with other biological findings: for example, dSPRINT predicts certain unknown DUF domains (DUF 1087 and DUF4074) to be DNA-binding domains. These unknown DUF domains are found in CHD and Hox proteins, respectively, both of which are indeed known to bind DNA.
Proteins are hard workers. They carry out all the actions that take place in your cells in order to keep you, as a whole, functioning. Yet, they can only do their jobs by interacting with the other proteins and molecules around them. Until we understand how these proteins interact with their surroundings, we can’t fully understand or predict protein function — or dysfunction, in the case of disease. Etzion-Fuchs et al. have created a model that can predict how protein domains interact with various ligands, without requiring labor-intensive crystallization techniques. In doing so, they have brought us far closer to understanding, harnessing, and manipulating these protein-ligand interactions: a key goal of molecular biology.
Etzion-Fuchs adds, “We are excited about using our predictions to assign novel functionalities to understudied proteins. Our dSPRINT framework can also pave a path to similar approaches beyond the human proteome. For instance, the ability to annotate ligand-binding properties of domains across the evolutionary spectrum may advance our understanding of the functional impact of the evolution of protein domain architectures. Our framework can also be combined with recent work of unsupervised learning of protein sequences to further improve protein function prediction from sequence.”
The dSPRINT framework, and all of its human protein domain binding predictions, are freely available at http://protdomain.princeton.edu/dsprint.