
Cecilia Panfil (CHM, 2022) and Alexandra Libby (PNI, GS)
Worldwide, approximately 250 million people have tested positive for the Hepatitis B virus (HBV). The virus infects the liver, causing severe damage when left untreated, such as chronic infection, liver fibrosis, liver cancer and cirrhosis. The likelihood of an adverse outcome or chronic illness is higher if the disease is contracted in childhood. Transmission can occur either through birth (i.e., the mother was infected) or close contact (e.g., sexual incourse or needle sharing for injectable drugs)1. HBV is a significant global health problem; overall, it is estimated that 650,000 people die each year from HBV related illnesses2.
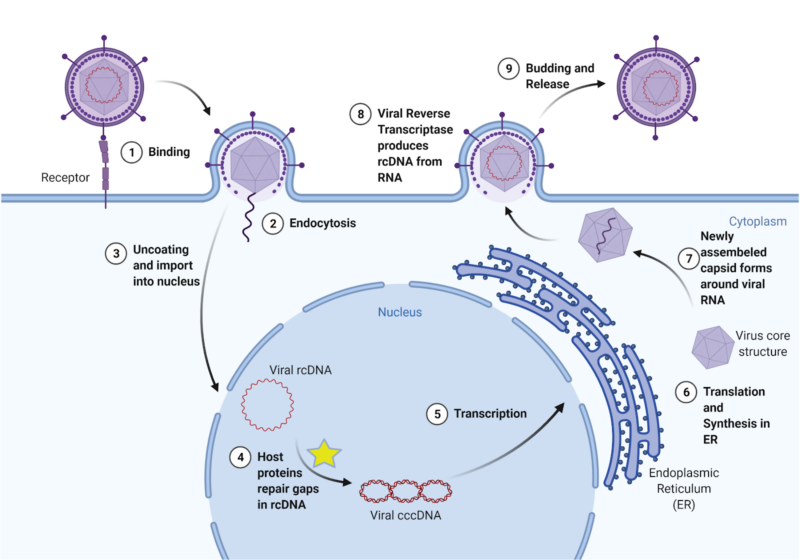
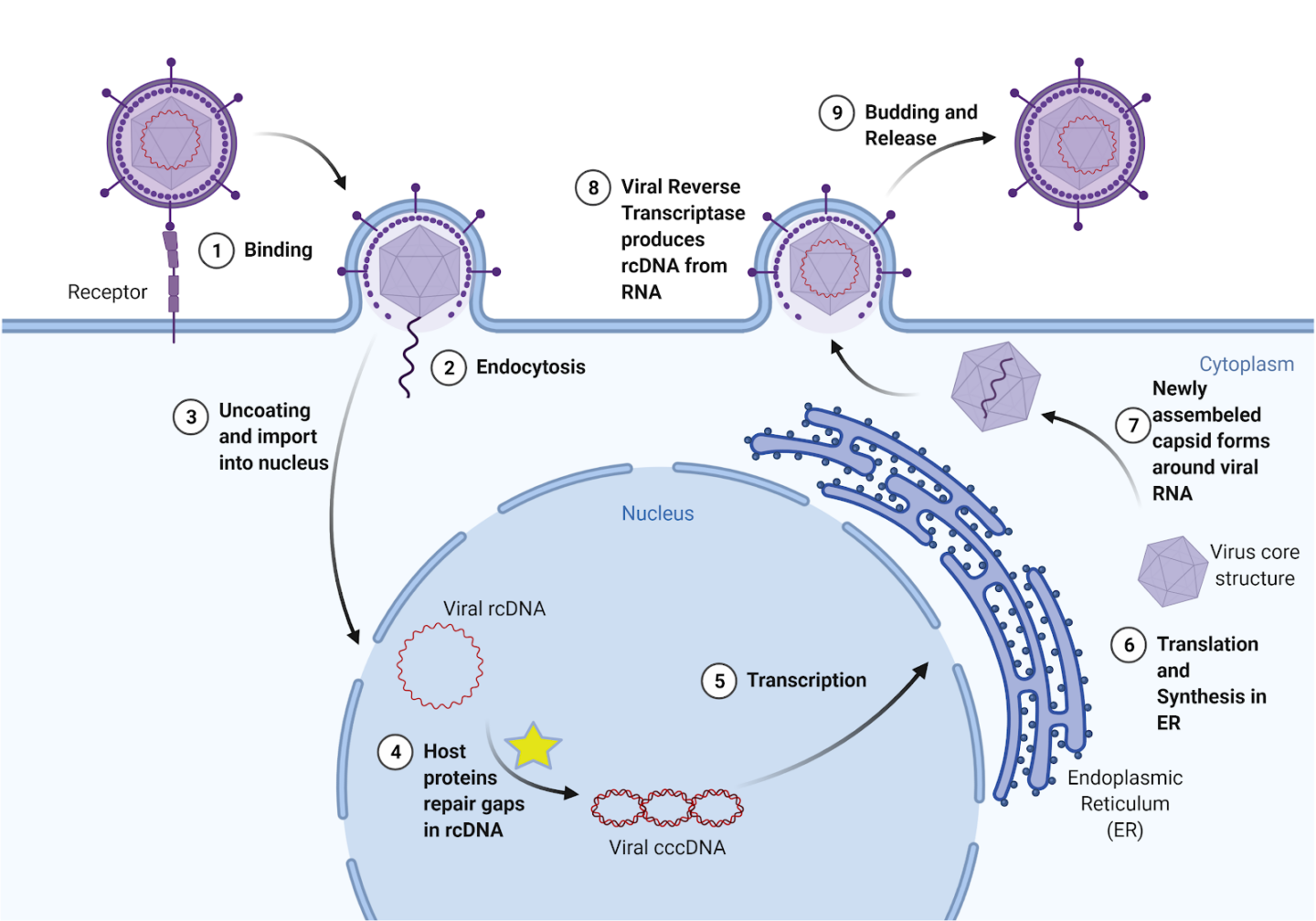
The virus acts by entering a liver cell and using the cell’s mechanisms to convert its broken, unreadable genetic material into a usable form, which then provides instructions for making new viruses (Figure 1). Currently, there are preventive vaccines, but there is no effective cure for chronic infection. One common treatment is called interferon therapy, which targets a viral polymerase, a protein that coordinates viral replication, for degradation. However, these treatments result in severe side effects and only reduce HBV in less than 10% of patients. Other available treatments have similarly low levels of effectiveness and patients often experience a resurgence of the virus (viral rebound) after stopping treatments, including patients who only had an acute HBV infection. Importantly, the transformation of HBV’s genetic material into a stable form (called cccDNA) plays a key role in HBV persistence. Therefore, effective HBV treatment requires understanding the molecular processes that stabilize the virus’s genetic matrix (i.e., cccDNA formation)2.
Recently, researchers in the Ploss lab at Princeton University uncovered proteins involved the virus’s deadly replication cycle. Specifically, in their recent publication, postdoctoral scholar Lei Wei and Professor Alexander Ploss discovered five key proteins that are necessary for the virus’s formation of cccDNA3. By uncovering the basic molecular machinery of HBV replication, Wei and Ploss have bettered our understanding of viral infection and laid a foundation for developing new, more effective treatments.
To continue reproducing, HBV needs cccDNA, or covalently closed circular DNA. Unlike human DNA, which is linear, cccDNA is continuously circular, with no clear opening or closing point. Because it is circular, it must be sealed shut, by the same bonds that hold every piece of DNA together. When HBV enters the cell, its DNA is relaxed and incomplete; this form is called relaxed, circular DNA, or rcDNA. Essentially, the virus’s DNA is “broken”; it is missing fragments and proper structure. If left as rcDNA, the virus’s genetic material will not be read, and therefore the code to produce all proteins necessary to build new viruses will not be transcribed. However, infected human cells treat this broken viral DNA as if it were its own cellular DNA and repair it, turning it into cccDNA. From then on, the cccDNA can be read by the infected cell to produce more viruses that go on to infect other cells. Because cccDNA is so stable, it is difficult to wipe out the virus once it is formed. Therefore, preventing the initial formation of cccDNA is key to finding a cure for HBV.
The virus is difficult to combat due to its similarities to native cellular machinery. Yet, these similarities led to a key insight about cccDNA formation. rcDNA is similar to the broken DNA the cell produces when replicating its own DNA. When the cell replicates DNA, it uses a protein called DNA polymerase to create new strands. But because of how the machinery works, there are initially gaps in the new DNA called okazaki fragments. Wei and Ploss hypothesized that the same proteins the cell uses to repair these okazaki fragments could be used to repair the virus’s rcDNA. They then tested these proteins in various experiments to see if they worked to turn rcDNA into cccDNA. First, they created a system to study cccDNA formation in a test tube. They created molecular constructs that resemble the HBV’s rcDNA and systematically tested which human proteins converted the rcDNA into cccDNA. They first did this in yeast cells, and then in HBV-infected human liver cells.
Wei and Ploss identified five proteins that the cell uses to repair rcDNA into cccDNA. All five were necessary; the removal of any one prevented cccDNA formation. Moreover, all five of the discovered proteins are involved in the repair of Okazaki fragments. One of the proteins they found was DNA Polymerase δ. They tested whether aphidicolin, a drug which inhibits this DNA polymerase, would also prevent cccDNA formation. To their excitement, aphidicolin greatly diminished cccDNA formation, making it a potential lead for new therapies.
Through their identification of these five key proteins, Wei and Ploss have uncovered an essential and previously unknown component of the mechanism of cccDNA formation during HBV infection. By expanding our understanding of cccDNA formation, a key factor in HBV’s resistance to standard treatments, Wei and Ploss are paving the way for the development of new, more effective therapies.
This article was published in Nature Microbiology on March 9, 2020. Please follow this link to view the full version.
References
- Schweitzer, A., Horn, J., Mikolajczyk, R. T., Krause, G. & Ott, J. J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. The Lancet, 386, 1546–1555 (2015).
- Nassal, M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut, 64, 1972–1984 (2015).
- Wei, L. & Ploss, A. Core components of DNA lagging strand synthesis machinery are essential for hepatitis B virus cccDNA formation. Nature Microbiology, 5, 715–726 (2020).