
Review written by Truc Do (CBE, post-doc)
Our microbial residents and their impacts
It has been estimated that there are at least as many bacterial cells in our bodies as there are our own cells1. The vast and diverse collection of bacteria and other microorganisms that live within and on us is known as the human microbiome. We are colonized with microorganisms from birth, but the structure (composition) of our microbial communities evolves throughout our lives2. In recent years, it has become increasingly apparent that human health is inextricably tied to the state of our microbiomes. For example, Crohn’s disease is an inflammatory bowel disease of increasing prevalence. Changes in the composition of the gut microbiota, as a result of diet and other environmental factors, have been associated with severe Crohn’s disease states3.
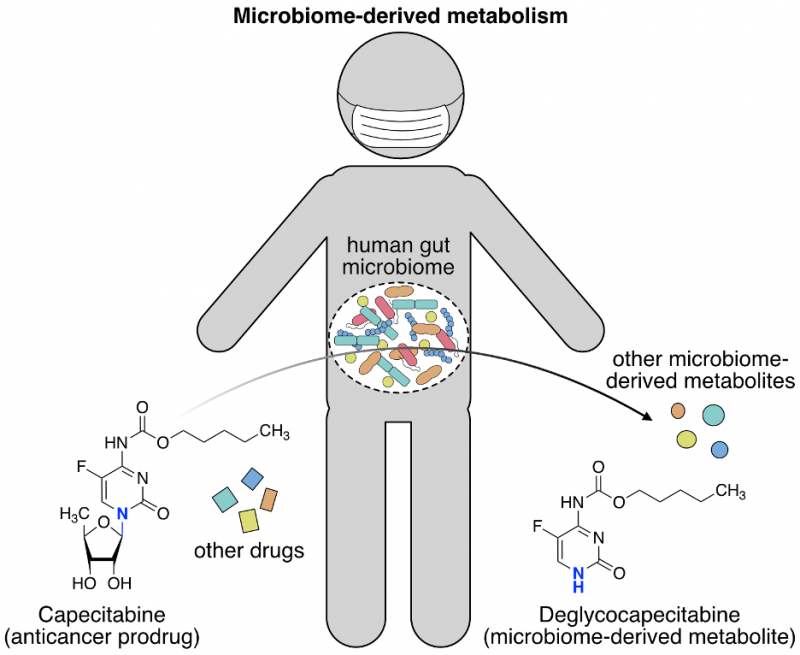
The makeup of our microbiome also impacts how we respond to drugs. Some drugs are administered to patients as an inactive precursor (prodrug) that is converted to the active form inside the body. The inactive form of the drug cannot exert the intended pharmacological effect unless it is converted to the active form. However, a prodrug that is prematurely activated can cause toxic side-effects or reduced bioavailability and efficacy. In some instances, the microbes that inhabit us are responsible for these adverse reactions. Such is the case with Levodopa, a drug used to treat the debilitating neurodegenerative disease Parkinson’s. The common gut bacteria Enterococcus faecalis was shown to metabolize Levodopa before it can enter the brain, thereby reducing the effective dose in patients4. To produce safe and efficacious therapeutics, it is critical that we understand the fate of drugs post-administration. The use of traditional microbiology techniques to assess drug metabolism in bacterial monocultures has provided a wealth of information on how specific organisms metabolize drugs. However, to truly anticipate how a drug may be transformed inside our bodies we need to determine what happens when a drug encounters the community of our resident microbes. In a major undertaking combining metabolomics, functional genomics, metagenomics, and mouse studies, the Donia group from Princeton’s Molecular Biology department describes a new experimental and computational framework to characterize how the human gut microbiome biochemically transforms clinically-important drugs5 (Javdan et al. Cell 2020). This and similar studies6 bring us closer to achieving tailored, personalized medicine that will improve treatment outcomes for patients.
Devising an optimized “broth” to recapitulate a patient’s microbiome
Studying the microbiome oftentimes requires first identifying a condition that supports growth of the bacteria. The standard lysogeny broth (LB) that is used worldwide to grow Escherichia coli, a common lab organism and the workhorse of molecular cloning, may be unsuitable as a growth medium for other bacteria. Indeed, many bacteria have never been successfully cultivated in the lab, leading to what is known as “the great plate count anomaly”7. Trying to grow a microbial community adds an additional layer of complexity: to the greatest extent possible, the medium should equally support the growth of all strains to prevent a few from overtaking the population. Javdan et al. set out to address this challenge and develop an ex vivo system for culturing donor-derived microbiomes by screening 10+ different media that are commonly used to grow gut microbes. The media were scored according to their ability to replicate the bacterial composition and diversity of the original donor fecal sample, as assessed through 16S ribosomal DNA (rDNA) sequencing and sequence variant comparison. A medium (mGAM, modified Gifu Anaerobic Medium) was identified that propagated a mixed culture most similar in structure to the original sample.
From there, the authors used this medium to cultivate the fecal-derived microbial community and test its ability to metabolize hundreds of oral drugs. High-performance liquid chromatography (HPLC), coupled with high-resolution mass spectrometry (HRMS) detection, was used to determine if the drugs were consumed to yield new molecules. This pilot screen revealed both previously known and novel metabolic activities. Some drugs were chemically converted to a new metabolite and others became depleted without concomitant detection of a product. Of those screened, 57 drugs, including the anti-inflammatory steroid hydrocortisone and the antiviral drug Famciclovir, appeared to be metabolized (MDM+, microbiome-derived metabolism positive). Drugs that were not biochemically transformed by the donor microbiomes were designated MDM- (microbiome-derived metabolism negative). The MDM+ drugs represented many distinct pharmacological classes, underscoring the pervasiveness of microbiome-derived metabolism.
Ex-vivo screens reveal hyper-variable MDM between patients
After establishing that their cultivation and screening methodologies were successful, as described above, the authors adapted the method to screen fecal samples from a cohort of 20 additional donors. To select a general media for cultivating the microbial communities from all donors, the Donia lab developed a statistical method to evaluate and maximize the diversity of a culture and the detection of metabolites. This method, coined Expected Number of Detectable Strains (ENDS), showed that mGAM mixed with a second media produced cultures with a desired profile.
The authors then measured the MDM capacity of these donor microbiomes (to test if the donor microbiomes can metabolize a panel of drugs) and explored whether there were differences between the MDM profile of each donor. The 20 donor fecal samples were cultivated in the selected media and screened against different drugs. Again, HPLC-HRMS was used to detect drug depletion and metabolite production. Some drugs were MDM- across all donor samples, whereas others were consistently MDM+ for all samples. Strikingly, several drugs showed a mixed MDM profile in that they were metabolized when incubated with a subset of donor fecal samples, but not all. An example of a mixed MDM-/MDM+ drug was the heart medication digoxin, which undergoes reduction to dihydrodigoxin in only 3 of the 20 donors. These results indicate that patient response to drugs, at least with respect to microbiome-derived metabolism of the drugs, can be highly variable.
Honing in on the specific bacteria and enzymes that metabolize two drugs
The authors went on to pinpoint the microbiome-derived genes that encode the enzymes which metabolize hydrocortisone and the chemotherapy drug capecitabine. Mechanistic information that explains how a drug is biochemically transformed is imperative for designing therapeutic strategies to circumvent unwanted metabolism in patients. From the MDM+ drugs that were identified in the screen, the authors chose to investigate (1) how capecitabine was deglycosylated (removal of a sugar group) to deglycocapecitabine and (2) how hydrocortisone was reduced to 20β-dihydrocortisone. These questions were answered using two different experimental approaches.
A computer-based search for proteins with similar amino acid sequences from an online protein database was employed to identify the enzymes that perform capecitabine deglycosylation, as similar sequence implies similar function. The human enzymes thymidine phosphorylase (TP) and uridine phosphorylase (UP) both deglycosylate a capecitabine-like molecule. Reasoning that bacterial proteins resembling TP and UP might also remove the sugar group of capecitabine, the Donia lab searched for these TP/UP-like proteins in bacteria that are present in the fecal sample. Fortunately, both TP and UP-like proteins were found in E. coli, a common inhabitant of the human gut. The TP-like protein was encoded by the gene deoA and the UP-like protein was encoded by the gene udp in E. coli. The authors deleted each of these genes in E. coli and compared the capacity of the mutant E. coli to metabolize capecitabine compared to non-mutated E. coli. A single udp gene deletion and simultaneous deletion of both the deoA and udp genes led to greater than 7-fold reduction in capecitabine deglycosylation, whereas deletion of deoA alone did not reduce deglycosylation levels. Therefore, udp appeared to specify the primary deglycosylating enzyme for capecitabine. However, both deoA and udp seemed to equally contribute to the deglycosylation of other fluoropyrimidines, a class of anticancer drugs to which capecitabine belongs.
An unbiased functional metagenomic screen was performed to uncover the enzymes that catalyzed ketone reduction of hydrocortisone. This second case was not amenable to computer-based searching as the types of enzymes that would perform hydrocortisone reduction are diverse and poorly characterized. Instead, the authors conceived a screening strategy to survey a large fraction of the donor metagenome and isolate clones that exhibit MDM activity on hydrocortisone. First, metagenomic DNA was prepared from a donor fecal sample and kilobase fragments of the DNA were cloned into an expression vector that could be replicated in E. coli. This library of 3 million clones was split into successively smaller pools to screen for pools that exhibited the desired MDM activity, ultimately leading to the isolation of a single clone. Gratifyingly, the clone encodes an enzyme which resembles a 20β-hydroxysteroid dehydrogenase, a type of enzyme that can perform hydrocortisone reduction.
In vivo assessment of MDM
The authors capped off this work with a mouse model experiment that assessed the in vivo relevance of the biochemical transformations of drugs observed ex vivo. As MDM activity depends on expression of the metabolizing enzymes, changes in their expression levels (due to growing the bacteria inside a living host vs. a test tube, for instance) have an impact on metabolite production. The authors returned to the example of capecitabine deglycosylation, but this time tested whether MDM transformation of capecitabine could be detected in the gastrointestinal tract of mice. Mice were initially treated with an antibiotic cocktail to flush out their native microbiome. Then, the mice were colonized with human donor fecal microbes and finally administered a dose of capecitabine. Compared to the non-colonized control mice, the human microbiome-colonized mice shed deglycocapecitabine in their feces, indicating that MDM deglycosylation also occurred in vivo. This finding showed that the microbial makeup of an organism impacts how it responds to treatment with a drug.
In summary, Javdan et al. carried out a multi-faceted study to understand how the human gut microbiome metabolizes drugs and discovered novel bacteria-driven biochemical transformations of common drugs. The study began with proof-of-principle experiments showing that an ex vivo system supports the growth of native-like gut microbial communities and enables detection of drug metabolites. In recent years, increasingly sophisticated sequencing technology has enabled researchers to map the individual species found within complex microbiomes. The field is now moving toward connecting broad descriptions of microbiome composition with detailed mechanistic information. Likewise, this study dug beyond detecting metabolites to explain how certain drugs are metabolized by microbes and why the patterns of metabolism may vary across patients. It has long been known that patients can respond very differently to drugs and some manifest negative side-effects with dire consequences. What has been less appreciated, but is gaining traction, is the role that our microbiome plays in how we react to drug treatments. A major finding of this study is that the variability in patient drug response owes in part to differences between patient microbiomes, specifically the bacteria that are present and the bacterial genes that are expressed. These findings highlight a critical need to consider and account for microbiome-drug interactions in the drug development pipeline, with an eye toward delivering safe and effective medicine that is tailored to each patient.
The original version of this article was published in Cell on June 25, 2020. Please follow this link to view the full version.
References
- Sender, R., Fuchs, S. & Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol 14, e1002533 (2016).
- Rodríguez, J. M. et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb Ecol Health Dis 26, 26050 (2015).
- Glassner, K. L., Abraham, B. P. & Quigley, E. M. M. The microbiome and inflammatory bowel disease. J Allergy Clin Immunol 145, 16–27 (2020).
- Maini Rekdal, V., Bess, E. N., Bisanz, J. E., Turnbaugh, P. J. & Balskus, E. P. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364, (2019).
- Javdan, B. et al. Personalized Mapping of Drug Metabolism by the Human Gut Microbiome. Cell 181, 1661–1679.e22 (2020).
- Zimmermann, M., Zimmermann-Kogadeeva, M., Wegmann, R. & Goodman, A. L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570, 462–467 (2019).
- Staley, J. T. & Konopka, A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol 39, 321–346 (1985).