
Review written by Laura A. Murray-Nerger (Molecular Biology, G6)
As primary and secondary school students, we learn that cellular organelles have specific functions. For example, the mitochondria is often called the “powerhouse” of the cell because it makes energy that drives other cellular processes. However, we often don’t learn about the multifaceted functions of these well-known organelles or learn about some of the less-well studied organelles, including the peroxisome. Moreover, as we learn about the functions of these organelles, it is easy to forget that they are filled with many proteins, each of which participates in a variety of functions. Importantly, these proteins do not work in isolation, but rather by interacting with each other, which creates a complex network of protein-protein associations that ultimately determine cellular fate. In their recent paper, the Cristea lab has built a computational platform that can be broadly used to assess the changes in protein-protein interactions in any biological context. They employ this newly developed tool to understand the protein-protein interactions that underlie alterations in mitochondrial and peroxisomal function during viral infection.
Tens of thousands of studies have aimed to understand the protein-protein interactions that underlie every aspect of cell biology. Among several approaches, many of which generate large datasets, a classical way to study these protein-protein interactions is through a technique known as immunoaffinity purification (IP), which allows the researcher to isolate a protein of interest through its affinity for an antibody that specifically recognizes that protein. Using this technique, other proteins that interact with the protein of interest will come along for the ride (i.e., the antibodies bind the protein of interest, and the protein of interest binds its interacting proteins), allowing all of these proteins to be identified and quantified by mass spectrometry. When this technique is performed across multiple biological conditions, massive datasets are quickly generated. Given the sheer scale of the data, it can be difficult to extract biological insights. Just imagine a researcher squinting at enormous spreadsheets, thousands of lines long, trying hopelessly to find the key but subtle elements that are biologically meaningful. Clearly, this manual needle-in-a-haystack analysis rapidly becomes tedious, overwhelming, and infeasible, especially for researchers with limited computational expertise.
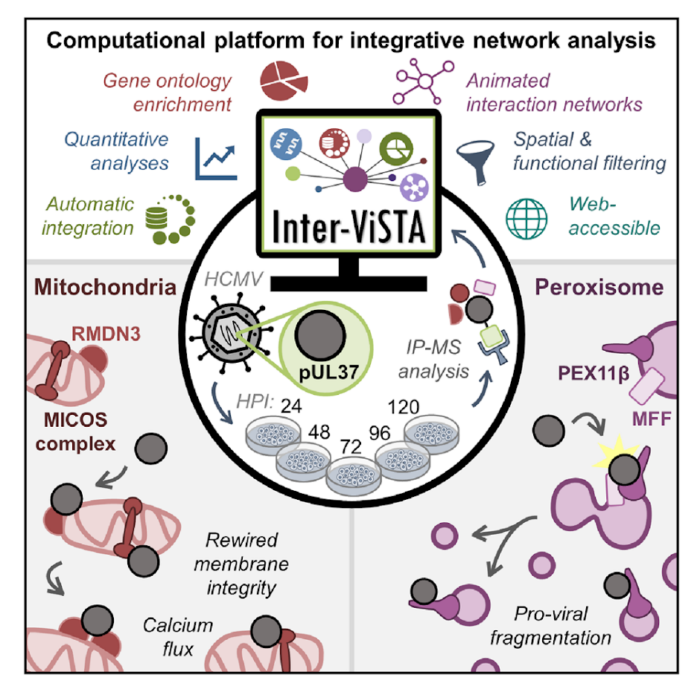
To address this labor-intensive data integration process, a team in the lab of Princeton’s Ileana Cristea, led by post-doc Joel Federspiel, graduate students Katelyn Cook and Michelle Kennedy, and undergraduate Samvida Venkatesh, developed a new web-based computational platform. This platform, named Interaction Visualization in Space and Time Analysis (Inter-ViSTA), seamlessly integrates user-supplied mass spectrometry data (amenable to multiple types of affinity-purification datasets) with online databases containing information about known protein-protein interactions, protein subcellular localization, protein function, and protein complex associations. Together, these data sources provide a more complete picture of how interactions with a particular protein of interest change over time. By putting all of these relevant pieces of information in a single location, Inter-ViSTA expedites the process of determining how protein-interaction patterns change over time and space and provides quantitative assessment of these trends. The Cristea lab has made this platform freely available (see publication for more details) in the hope that it will serve as a “research accelerator” to speed the process of mining proteomic data and generating biological hypotheses.
One area of particular interest for understanding the complexities of protein-protein interactions is the field of viral infection. New viruses, like SARS-CoV2, are continually evolving; understanding the protein-protein interactions that underlie the replication of these viruses can provide critical information to develop novel treatment strategies. To this end, the authors leveraged Inter-ViSTA to investigate a major focus of the Cristea Lab: understanding how protein-protein interactions are altered during viral infection and how these interactions impact organelles and their functions. They focused on the prevalent human pathogen human cytomegalovirus (HCMV), which infects 50-90% of the population. Despite being a dangerous pathogen for immunocompromised individuals and the leading cause of virally induced birth defects, there is no vaccine nor permanent treatment for HCMV. Better defining the complex replication cycle of this virus can aid in the development of novel therapeutic interventions. One hallmark of HCMV infection is that it induces a dramatic reorganization and restructuring of all organelles that are required for virus production. For example, HCMV proteins interacting with host proteins induce the reorganization of the Golgi and the ER into an entirely new organelle, the viral assembly complex, where viral particles complete their maturation and assembly. Another key organelle reorganization is the fragmentation of mitochondria and peroxisomes. This means that host mitochondria change from having a long, tubular morphology to a fragmented morphology and peroxisomes split into two populations (irregular large peroxisomes and very small round peroxisomes). While these perturbations are broadly linked to pro-viral changes including alterations to cellular metabolism (mitochondria) and inhibition of the immune response (peroxisomes), understanding the mechanisms that drive fragmentation of these organelles is still an ongoing area of research.
In this study, Federspiel and colleagues focus on the role of a critical viral protein, pUL37x1, which is known to localize to mitochondria and peroxisomes during infection. Prior to this study, the function of pUL37x1, particularly at late stages of infection, was poorly understood. Federspiel et. al. employed Inter-ViSTA followed by functional analyses (microscopy to determine protein localization and organelle morphology, CRISPR-mediated knockout of important proteins, viral titer assays to assess the amount of virus produced) to demonstrate that the viral protein pUL37x1 has roles in both mitochondrial and peroxisomal fragmentation during HCMV infection. By leveraging the Inter-ViSTA interface to integrate changes in protein associations over time with information regarding protein function, they discovered that pUL37x1 associates with a complex of core mitochondrial proteins that are essential for maintaining mitochondrial structure. This observation led the authors to hypothesize that these protein-protein interactions may be a missing link in defining the mitochondrial fission induced by pUL37x1. The authors also define protein-protein interactions with pUL37x1 that are required for its mediation of peroxisomal fragmentation and show that pUL37x1 is localized to the small, fragmented peroxisomes and is excluded from enlarged peroxisomes. These findings suggest that pUL37x1 plays a pro-viral role in generating the dual peroxisome populations during infection.
Through this work, Federspiel et al. develop a new, user-friendly platform for integrating large, protein-based datasets and biological knowledge. The authors show the utility of this platform through their discoveries about pUL37x1 and its role in peroxisomal fragmentation during HCMV infection. Inter-ViSTA serves as an exciting launching point for streamlining the protein interaction analysis process and facilitating new biological advances.
This original article was published in Cell Reports on July 28, 2020. Please follow this link to view the full version.
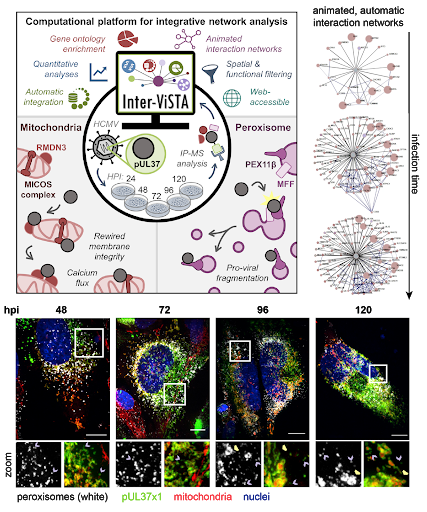
Figure 1. Inter-ViSTA is a powerful tool to analyze protein-protein interaction and tease apart biological meaning. Interactive networks allow visualization of changes in interactions over time (upper right). The authors use a combination of techniques, including microscopy, to illustrate the role of the viral protein pUL37x1 in regulating peroxisomal and mitochondrial morphology (model, upper left; representative images, bottom, hpi is hours post infection). Figures adapted from Federspiel, et al.