
Review written by Kimberly Sabsay (QCB, G2)
Individual cells go through life cycles, in which they grow and prepare for cell division. Similarly, in certain bacteria, groups of cells go through a synergistic cycle involving the formation and disassembly of biofilms. Biofilms are essentially groups of cells that surround themselves in layers of self-made extracellular matrix proteins and polysaccharides. A biofilm is a way for cells to protect themselves from dislocation or death by predation and are present in both beneficial and pathogenic bacterial microbiomes. The sticky extracellular matrix is a structure that pathogens such as Vibrio cholerae (the bacteria that causes Cholera) can hide in to avoid detection by immune cells and thus increase their virulence. In this way, biofilms essentially provide protective armor for bacterial cells that inhibits our ability to fight pathogenic infections.
Scientific understanding of Vibrio cholerae’s biofilm life cycle has been previously focused on the first stage, biofilm formation, while little has been done to understand the final stage, biofilm dispersal (Figure 1A). Dispersal of pathogenic biofilms lessens the virulence and alleviates the severity of infection. Understanding how biofilm dispersal occurs in its natural life cycle can direct our attention to viable and novel therapeutics for treating cholera and other pathogenic bacterial infections. Andrew Bridges and Chenyi Fei, graduate students at Princeton University advised by Bonnie Bassler, have recently paved the way to understanding how biofilm dispersal is regulated in their recent work entitled “Identification of signaling pathways, matrix-digestion enzymes, and motility components controlling Vibrio cholerae biofilm dispersal” (PNAS, 2020).
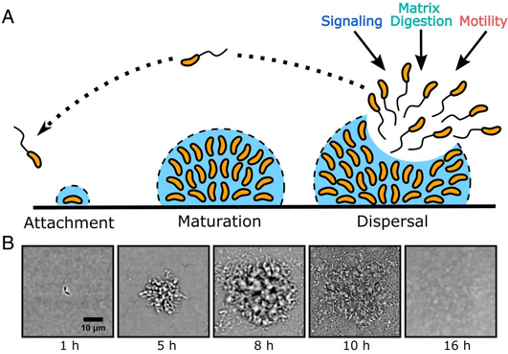
Utilizing a brightfield imaging technique they previously developed, Bridges and colleagues are able to watch V. cholerae’s biofilm life cycle in its entirety, in real-time. In wild-type (WT) V. cholerae, i.e. the naturally occurring bacterial strain, biofilm formation reaches peak size/maturation after 8-9 hours. Immediately following, dispersal begins and is completed by 12-13 hours post inoculation with only about 6% of the peak biofilm remaining (Figure 1B).
The team constructed a library of about 7,000 V. cholerae mutants using transposon mutagenesis, a technique by which individual genes are disrupted in an organism’s genome. They used the same imaging technique, harnessing the power of brightfield microscopy, to monitor each of the mutants individually. To focus their study on biofilm dispersal, they selected and identified mutants which appeared to form a mature biofilm as successfully as the WT V. cholerae strain, but lacked the ability to perform complete biofilm dispersal. Since the mutants’ behavior clearly differed from the WT strain in some aspect pertaining to biofilm dispersal, these mutants provide evidence as to what specific portions of the genome are crucial for regulation.
Of the 47 selected mutants, the genes identified as important for biofilm dispersal belonged to three categories: signal transduction, matrix degradation, and motility. For each of the identified genes of interest, mutants were made with in-frame deletions (resulting in a complete knock-out of that gene). These mutants were then imaged to confirm that biofilm dispersal was significantly hindered compared to WT V. cholerae, as suggested by the initial imaging screen.
Gene regulation
The mutant that inhibited biofilm dispersal most dramatically contained a disruption in the gene named vc1639, which encodes for a protein called histidine kinase (HK). This protein is responsible for sending sensory information to response regulators (RRs), which in turn control gene expression. Complete deletion of this gene, vc1639, resulted in 80% more biofilm formation at its peak with almost zero dispersal evident after 16 hours, compared to WT V. cholerae. This is evidence that vc1639 is crucial for successful biofilm dispersal.
Matrix digestion
Other selected mutants disrupted both the LapG gene, which codes for a protein that degrades outer-membrane adhesive proteins (LapG), and the RbmB gene, which codes for a protein that degrades the vibrio polysaccharide (VPS) component of the biofilm matrix crucial for biofilm formation. The researchers intentionally created a knock-out mutant, ΔlapG, which did not possess the lapG gene. This mutant displayed a delay in dispersal with 55% of the peak biofilm still remaining after 16 hours. By genetically attaching a light producing operon (collection of genes) to the VPS gene, researchers can monitor the expression of VPS by quantifying the amount of light produced. In this way, they found that the ΔlapG mutant produced similar levels of VPS as compared to the WT. Deletion of lapG thus does not directly alter the expression of VPS, revealing that LapG does not inhibit matrix formation but functions later on in the biofilm life cycle during matrix degradation.
In previous research, the LapG mechanism has been found to remove two adhesion proteins from the outer membrane after detecting a drop in concentration of a signaling molecule. The Princeton team’s results for V. cholerae are consistent with this mechanism; without LapG, as in the ΔlapG mutant, these two adhesion proteins do not get removed and thus continue to bind the cells to the matrix, prohibiting them from dispersing. Bridges et al.’s results confirm that the Lap regulatory pathway directly controls biofilm dispersal.
Additionally, the knock-out mutant ΔrbmB also displayed a delay in onset of dispersal; the majority of the biofilm remained after 16 hours for this mutant strain. The authors hypothesized that, without RbmB, VPS within the biofilm would not be degraded. This, in turn, would prevent dispersal. By staining non-dispersed biofilms with a red dye that binds specifically to VPS, the researchers visually quantified the amount of VPS present. Since they had previously established that the dispersal defect from the ΔlapG mutant was independent of VPS, Bridges and Fei compared the ΔrbmB mutant to the ΔlapG mutant and found that the ΔrbmB mutant had six times the amount of VPS present. This is consistent with the fact that the ΔlapG mutant still retained the RbmB functionality to degrade VPS while the ΔrbmB mutant could not degrade VPS. Thus, it can be concluded that RbmB functionality is also critical for biofilm digestion and dispersal.
Cell motility and reorientation
A particular dispersal-defective mutant identified in the screen disrupted a required chemotaxis gene, cheY3. cheY3 is responsible for the non-random direction-specific movement of V. cholerae cells. While less defective than some of the previous mutants, this knock-out mutant, ΔcheY3, still retained about 21% of the peak biofilm after 16 hours.
Each V. cholerae cell contains a single propelling “tail” called a polar flagellum that directs movement by rotation. When CheY3 is activated, it binds to the flagellum and induces a switch from counterclockwise rotation (CCW), allowing cells to travel in smooth straight paths, to clockwise rotation (CW) rotation to reorient the direction of travel. The knock-out mutant, ΔcheY3, lacks the ability to reorient directionality of travel and the cells are required to move only in straight directions via CCW rotation. The researchers created an additional mutant that locks the flagellar “tail” in a CW rotation (cheY3*) and compared it to the original ΔcheY3 mutant to investigate whether the cheY3 defect is specific to missing a CW to CCW switch.
The cheY3* mutant produced cells that were only propelled via CW flagellar rotation and thus could not swim in smooth straight lines. Interestingly, the cheY3* mutant retained the WT dispersal ability despite the lack of chemotactic ability. This suggests that the dispersal defect seen in the originally selected ΔcheY3 mutant is not due to the lack of chemotaxis, but rather a direct result of the inability to reorient travel direction.
To confirm this, the researchers measured the turning frequency of the ΔcheY3 and cheY3* mutants. Compared to the WT strain turning frequency of about one turn every 3 seconds, the ΔcheY3 mutant barely turned at all with less than one turn every 40 seconds, consistent with the forced straight-line travel due to the constant CCW flagellar rotation. On the other hand, the cheY3* mutant turned once every 0.5 seconds, consistent with the expected lack of smooth and straight traveling and the constant reorientation due to the CW flagellar rotation.
The results imply that the especially low turning frequency of the ΔcheY3 mutant is directly responsible for the limited biofilm dispersal ability. They propose that the cells need to be able to turn in order to navigate through the dense biofilm matrix in order to escape. It has been shown that in other bacteria, straight-swimming mutants cannot travel through mesh-like media very well compared to their WT counterparts.
Conclusion
The recent work conducted by Andrew Bridges, Chenyi Fei, and Bonnie Bassler on Vibrio cholerae biofilm dispersal has given us our first insights into the final stage of the biofilm life cycle. The most important aspects controlling biofilm dispersal are a) the regulatory system controlled by the gene vc1639, b) the matrix digestion proteins LapG and RbmB, and c) the ability for cells to reorient their swimming directionality. Not only does this fill in the remaining gaps in our understanding of the complete biofilm life cycle, but it also elucidates key components that regulate biofilm dispersal that can now be targeted to combat virulence factors. This could reduce infection severity and even prevent transmission of bacterial pathogens like V. cholerae.
“Going forward we are very excited to study the mechanism of Vc1639 function, which we have since named DbfS. We want to know what signals are transmitted by the receptor and how these signals are pertinent to the biofilm life-cycle of V. cholerae. Ultimately we would like to be able to control the activity of Vc1639 with new small molecules that could potentially be used as therapeutics.” – Andrew Bridges
This original article was published in Proceedings of the National Academy of Sciences on December, 22, 2020. Please follow this link to view the full version.